
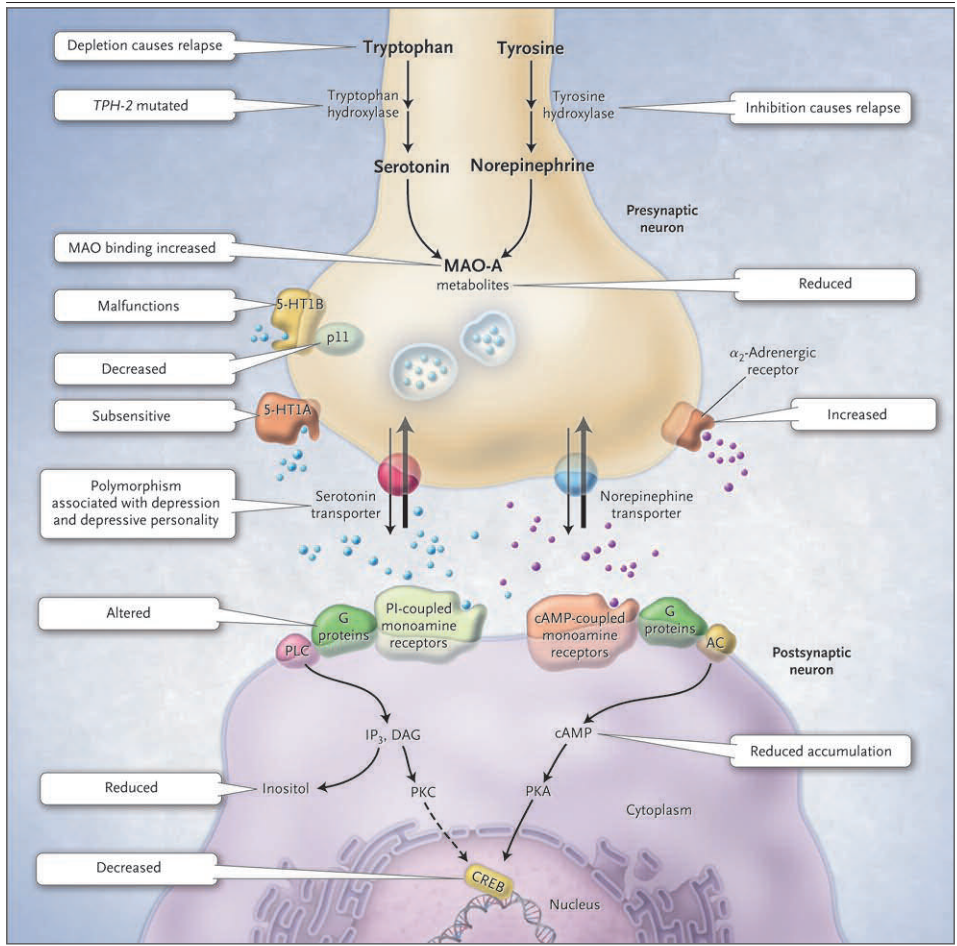
Figure 1 (facing page). The Monoamine-Deficiency Hypothesis Extended. The monoamine hypothesis of depression postulates a deficiency in serotonin or norepinephrine neurotransmission in the brain. Monoaminergic neurotransmission is mediated by serotonin (5-hydroxytryptamine 1A [5-HT1A] and 5-hydroxytryptamine 1B [5-HT1B]) or norepinephrine (noradrenaline) released from presynaptic neurons (serotonergic neuron, shown on the left side, and noradrenergic neuron, shown on the right side [condensed virtually]). Serotonin is synthesized from tryptophan, with the first step in the synthetic pathway catalyzed by tryptophan hydroxylase; norepinephrine is synthesized from tyrosine, with the first step catalyzed by tyrosine hydroxylase. Both monoamine transmitters are stored in vesicles in the presynaptic neuron and released into the synaptic cleft, thereby affecting both presynaptic and postsynaptic neurons. Cessation of the synaptic action of the neurotransmitters occurs by means of both reuptake through the specific serotonin and norepinephrine transporters and feedback control of release through the presynaptic 5-HT1A and 5-HT1B regulatory autoreceptors for serotonin and the α2-noradrenergic autoreceptors for norepinephrine. Monoamine oxidase A (MAO-A) catabolizes monoamines presynaptically and thereby indirectly regulates vesicular content. The protein p11, which interacts with 5-HT1B receptors, increases their function. Postsynaptically, both serotonin and norepinephrine bind two kinds of guanine nucleotide triphosphate–binding protein (G protein)–coupled receptors: cyclic AMP (cAMP)–coupled receptors, which activate adenylate cyclase (AC) to generate cAMP, and phosphatidylinositol (PI)–coupled receptors, which activate phospholipase C (PLC). PLC generates inositol triphosphate (IP3) and diacylglycerol (DAG); cAMP activates protein kinase A (PKA), and IP3 and DAG activate protein kinase C (PKC). The two protein kinases affect the cAMP response element–binding protein (CREB). Findings in patients with depression that support the monoamine-deficiency hypothesis include a relapse of depression with inhibition of tyrosine hydroxylase or depletion of dietary tryptophan, an increased frequency of a mutation affecting the brain-specific form of tryptophan hydroxylase (TPH-2), increased specific ligand binding to MAO-A, subsensitive 5-HT1A receptors, malfunctioning 5-HT1B receptors, decreased levels of p11, polymorphisms of the serotonin-reuptake transporter associated with depression, an inadequate response of G proteins to neurotransmitter signals, and reduced levels of cAMP, inositol, and CREB in postmortem brains.
図1(見開きページ)。モノアミン欠乏症仮説・拡大版。
うつ病のモノアミン仮説は、脳内のセロトニンまたはノルエピネフリン神経伝達の欠乏を仮定している。モノアミン神経伝達は、シナプス前細胞から放出されるセロトニン(5-ヒドロキシトリプタミン1A[5-HT1A]、5-ヒドロキシトリプタミン1B[5-HT1B])またはノルエピネフリン(ノルアドレナリン)により媒介されている(左側がセロトナー神経、右側がノルアドレナリン神経[縮図])。セロトニンはトリプトファンから合成され、その合成経路の第一段階はトリプトファン水酸化酵素によって触媒され、ノルエピネフリンはチロシンから合成され、その第一段階はチロシン水酸化酵素によって触媒される。両者ともシナプス前神経細胞内の小胞に貯蔵され、シナプス間隙に放出され、シナプス前神経細胞とシナプス後神経細胞の両方に影響を与える。神経伝達物質のシナプス作用の停止は、セロトニンおよびノルエピネフリン輸送体による再取り込みと、セロトニンについてはシナプス前の5-HT1Aおよび5-HT1B制御自己受容体、ノルエピネフリンについてはα2神経性自己受容体による放出のフィードバック制御によって行われる。モノアミン酸化酵素A(MAO-A)はシナプス前細胞でモノアミンを異化し、それによって間接的に小胞内含有量を調節している。5-HT1B受容体と相互作用するタンパク質p11は、5-HT1B受容体の機能を高める。セロトニンとノルエピネフリンは、シナプス後において、アデニル酸シクラーゼ(AC)を活性化してcAMPを生成するサイクリックAMP(cAMP)共役型受容体と、ホスホリパーゼC(PLC)を活性化してPI共役型受容体の2種類のグアニンヌクレオチド三リン酸結合タンパク質(Gタンパク質)共役受容体に結合する。PLCはイノシトール三リン酸(IP3)とジアシルグリセロール(DAG)を生成し、cAMPはプロテインキナーゼA(PKA)を活性化し、IP3とDAGはプロテインキナーゼC(PKC)を活性化する。この2つのプロテインキナーゼは、cAMP応答エレメント結合タンパク質(CREB)に影響を与える。モノアミン欠乏症仮説を支持するうつ病患者の所見としては、チロシン水酸化酵素の阻害や食事性トリプトファンの欠乏によるうつ病の再発、トリプトファン水酸化酵素の脳特異的形態(TPH-2)に影響を与える変異の頻度の増加、MAO-Aに結合するリガンド特異性の増加などがある。ほかにも、5-HT1A受容体の感受性低下、5-HT1B受容体の機能低下、p11の減少、うつ病に関連するセロトニン再取り込みトランスポーターの多型、神経伝達物質シグナルに対するGタンパク質の不十分な反応、死後脳におけるcAMP、イノシトール、CREBレベルの低下などがある。
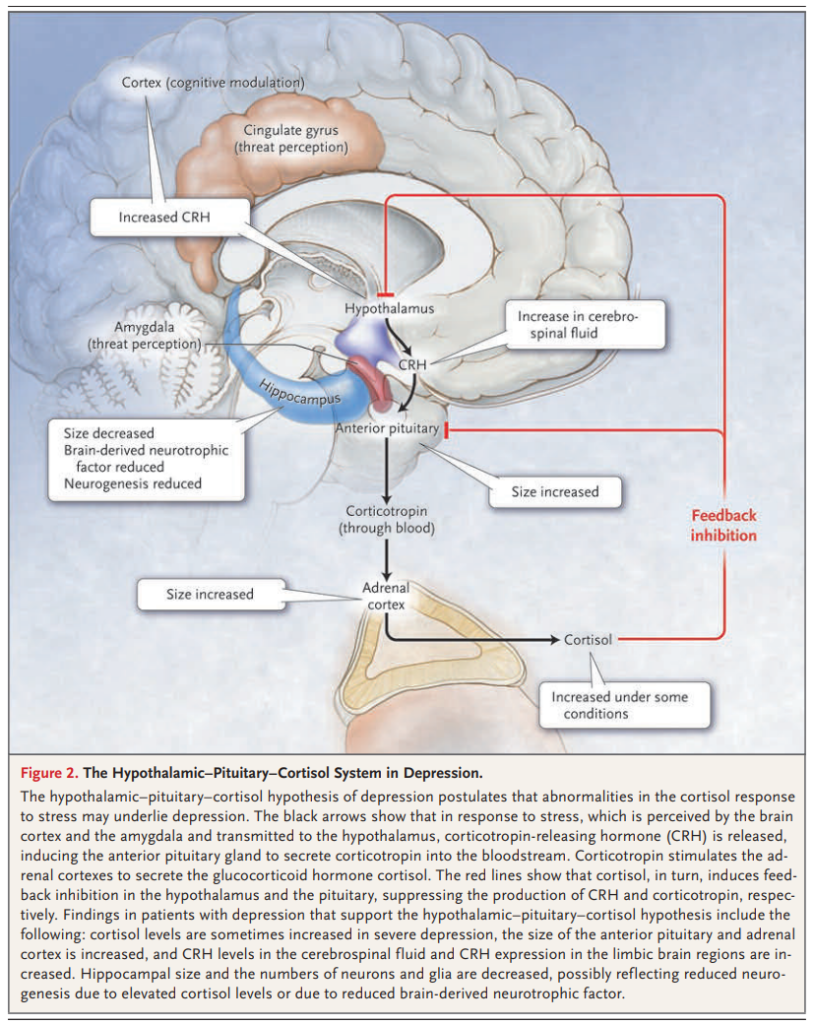
図2.うつ病における視床下部-下垂体-コルチゾール系。
うつ病の視床下部-下垂体-コルチゾール仮説は、ストレスに対するコルチゾール反応の異常がうつ病の背景にある可能性を仮定している。黒い矢印は、大脳皮質と扁桃体で感知されたストレスが視床下部に伝わると、コルチコトロピン放出ホルモン(CRH)が放出され、下垂体前葉からコルチコトロピンが血中に分泌されることを誘導している。コルチコトロピンは、副腎皮質を刺激してグルココルチコイドホルモンであるコルチゾールを分泌させる。赤い線は、コルチゾールが、視床下部と下垂体においてフィードバック抑制を引き起こし、それぞれCRHとコルチコトロピンの産生を抑制していることを示している。視床下部-下垂体-コルチゾール仮説を支持するうつ病患者における所見としては、重度のうつ病ではコルチゾールレベルが時に上昇し、下垂体前葉と副腎皮質のサイズが増大し、脳脊髄液中のCRHレベルと辺縁系脳領域でのCRH発現が増大することなどがあげられる。海馬の大きさ、神経細胞とグリアの数は減少しており、これはコルチゾールレベルの上昇による神経新生の減少、あるいは脳由来神経栄養因子の減少を反映していると考えられる。
